Initial trials and tribulations towards understanding site-directed mutagenesis effects on Staphylococcus aureus ketopantoate reductase
by Rishabh Jaryal
The enzyme Ketopantoate Reductase (KPR) catalyzes the NADPH-dependent reaction of ketopantoate to pantoate, which is ultimately converted into coenzyme A. Coenzyme A is an essential part of metabolic pathways to create energy. Pathogenic bacteria, not humans, rely upon KPR to power basic cellular functions and it has been identified as target for the development of new antibacterial drugs. A wealth of mutagenic information from previous studies is available for KPR from E. coli, which have revealed ASN98 to be important for KPR function1. Whether or not this amino acid plays an equally important role in S. aureus remains unknown. Presented here are preliminary results of an ongoing site-directed mutagenesis experiment in which ASN97 KPR (S. aureus) was mutated to GLN97 to better understand the important role that this amino acid plays. Mutagenesis was successfully performed by mutagenic-primer based PCR as verified by restriction enzyme digestion and DNA sequencing. Limited time in the classroom research lab prevented further work, but this research can be continued by a future student.
Keywords: site-directed mutagenesis, ketopantoate reductase, S. aureus
Introduction
Metabolic pathways play a critical role in energy absorption, production, and usage. A pathway of central importance to energy production involves ketopantoate reductase (KPR), which catalyzes the NADPH-dependent reduction of ketopantoate to pantoate[1, 2]. KPR is one of several enzymes in the pathway that synthesizes pantothenic acid (vitamin B5), which is the substrate for coenzyme A (CoA). CoA is an essential cofactor in energy metabolism, therefore obtaining or synthesizing pantothenic acid is important for growth and survival or an organism. Mammals, including humans, synthesize CoA from dietary sources of pantothenic acid. However, for other organism such as prokaryotic bacteria, the synthesis of pantothenic plays a larger role in CoA production and energy metabolism. Because of this unique disparity, the pantothenate synthesis pathway has been identified as a candidate target for the development of new antibacterial drugs[3, 4].
Studying the structure and function of KPR may accelerate the drug design process for new antibiotics. Ciulli et al. (2007) investigated which amino acids are important for KPR function in Escherichia coli (E. coli) through a process called site-directed mutagenesis (SDM). (The interested reader is directed to Carter et al. [5] for a review on SDM). They identified key residues that are important for KPR’s function [6]. One of these residues is asparagine 98 (N98), which is located in the enzyme’s active site and helps the enzyme bind the substrate. When mutated, KPR was not able to function as well as the nonmutated (wildtype) KPR revealing this amino acid’s importance.
However, less information mutagenic information exists for KPR found in pathogenic Staphylococcus aureus (S. aureus), which caused 20,000 deaths in 2017 ([7]). One study of S. aureus KPR showed, contrary to E. coli, that KPR may exists as a dimer and functions using kinetic Cooperativity [8]. This study also showed that the 97th residue in KPR (S. aureus) is in the relative position as N98 for E. coli KPR. However, this study did not further investigate this amino acid by mutagenesis. We hypothesize that residue N97 of S. aureus KPR functions similar to N98 of E. coli because it is located in the same positive relative to the active site. Studying this corresponding amino acid in S. aureus KPR is important to better understanding how it functions and may aid the development of new antibiotic drugs.
To better understanding ASN97’s role in S. aureus KPR, we set out to perform a site-directed mutagenesis (SDM) experiment in which N97 was mutated to glutamine. The end goal is to perform structural and functional studies. In this article we report the preliminary steps of this experiment, which was conducted as part of a course-based research project in the Biological Sciences Division. An initial trial, which had required repeating due to technical user-error, was discussed along with a description of the necessary troubleshoot steps leading to the creation of an N97Q mutation in the KPR gene. This mutation was performed on the DNA level and verified by restriction enzyme digestion and DNA sequencing.
Materials and Methods
Structural Analysis. The protein structure corresponding to S. aureus KPR (PDB ID 4YCA) was investigated using a molecular graphics software program, UCSF Chimera [9].
Primer design for mutagenic PCR. Mutagenic primers were developed using the software programs SDM-Assist [10] and SnapGene (from Insightful Science; available at snapgene.com). SDM-assist was used to develop candidate primers and SnapGene was used to visualize and verifying the best primers for mutation verification with Restriction Enzyme digestion. A forward primer (5’-TAGCCCAACAGGGTTATGGCCAACTCGAACATATTCCA-3’) and reverse primer (5’-TCGAGTTGGCCATAACCCTGTTGGGCTAAAATGATGAG-3’) were developed to create a missense mutation (Q97N) in the KPR gene. A silent mutation was also created to create a new restriction enzyme site for the restriction endonuclease, MscI.
PCR-based mutagenesis. The template plasmid used in the mutagenic PCR reaction was a pSX3 expression vector containing the full-length gene for S. aureus KPR (residues 1-286). This plasmid was kindly provided by Dr. Zachary Wood (Department of Biochemistry and Molecular Biology, University of Georgia). PCR-based mutagenesis was performed using the QuikChange Lightning Mutagenesis Kit™ (Agilent) by following the manufacturer’s provided protocol. Briefly, a 50µL µL PCR reaction was set up recommendation protocol using 100ng of template plasmid and primers at working concentration of 1µM. A control PCR reaction provided with the kit was also performed according to the manufacturer’s recommended protocol. Both reactions underwent thermal cycling with the following segments: Segment 1) Initial denaturation at 95°C for 2 mins, Segment 2) 18 cycles of 95°C for 20 sec., 60°C for 10 sec., and 68°C for 30 sec./kb of plasmid length, Segment 3) 68°C for 5 min.
Gel Electrophoresis of PCR product. A 1.0% agarose gel in TAE (Tris, Acetic Acid, and EDTA) buffer was used for all DNA analysis. Ethidium bromide was added to the gel solution at a final concentration of .2µg/mL to visualize DNA upon UV imaging.
Transformation of plasmid. E. coli was transformed with the PCR reaction and transferred to LB-agar plates according to the manufacturer’s instructions found in the QuikChange Lightning Mutagenesis Kit™ (Agilent). Briefly, LB-agar plates were prepared with ampicillin at a final concentration of 50µg/mL to select for cell that have taken up the control PCR reaction. LB-agar plates were prepared with kanamycin at a final concentration of 50µg/mL to select for cells that have taken up the experimental PCR reaction. After the PCR reactions were finished, the control and experimental sample were each treated with 2µL of Dpn1 restriction endonuclease in order to digest parental plasmid. 10µL of the Dpn1-treated reaction was mixed with 45µL of XL10-Gold Ultracompetent cells and incubated on ice for 30min. The samples were heat-pulsed at 42°C for 30 sec. and returned back to ice. NZY+ broth was added to each transformation solution and placed in a shaking incubator at 37°C for 1 hour at 225-250 rpm. Each transformation solution was transferred to LB-agar plates containing the appropriate antibiotic and incubated at 37°C for 16 hours.
Plasmid Purification. Plasmids from a colony were purified using the QiaPrep Plasmid MiniPrep Kit™ (Qiagen) by following the manufacturer’s suggested protocol. Briefly, culture tubes were made with 5 mL LB broth containing either Kanamycin or ampicillin as selection criteria. A culture tubes was inoculated with a colony and placed in a shaking incubator at 250RPM for 16 hours at 37°C. The entire culture volume was harvested for plasmid purification by centrifugation for 2min. at 5,000rpm. The cell pellet underwent the manufacturer’s protocol for cell resuspension, alkaline lysis, and neutralization. Plasmid DNA was eluted from spin columns using molecular grade water.
DNA sequence alignment. Purified plasmid samples were submitted for DNA sequencing to GeneWiz (For additional information about GeneWiz, the reader is directed to https://www.genewiz.com/en,). DNA sequences were analyzed using the SnapGene.
Results and Discussion
Chimera structure of S. aureus KPR. This research project is a part of a course-based research project in the course, BIOL3110L: Basic Skills in the Laboratory. The purpose of our research project is to better understand S. aureus KPR suing site directed mutagenesis. N98 was previously identified in mutagenic studies as critical for KPR function in E. coli. However, the role that this residue plays in S. aureus remains unknown. The long-term objective of this research project is to mutate the corresponding amino acid (N97) in S. aureus KPR.
Prior to creating any mutation, we analyzed the structure of KPR to better understand what mutation we were interested in doing. This was achieved by investigating the published crystal structure of S. aureus KPR (PDB ID 4YCA) with the software program UCSF CHIMERA. The first goal was to identify the amino acid in S. aureus that corresponds to N98 in E. coli. We achieved this by comparing the S. aureus model with the E. coli model relevant in E. coli, (PDB ID 2OFP). We identified the 97th residue in KPR S. aureus to be an asparagine and also located in the same relative location as N98 in E. coli. Therefore, we identified this residue as our target amino acid for mutation.
The next step was to analyze the possible effects of mutating N97 to Q97. We were able to analyze the effects of the mutation by performing an in-silico mutation with the UCSF CHIMERA program’s Rotamer function. The mutant was compared with wildtype KPR (wtKPR). Comparing wtKPR (Figure 1A) with the mutated molecule (Figure 1B), we can predict what may happen to the overall molecule from the mutation. Looking at the different amino acid, the original asparagine has an ethyl group, which would get changed to a propyl group with substitution of glutamine. The interaction with NADP+ varies with both amino acids. Looking at Figure 1A, there is only one hydrogen bond between asparagine and NADP+. This bond exists between asparagine and a hydroxyl group of NADP+. Whereas in Figure 1B, there are two hydrogen bonds between glutamine and NADP+. One bond exists between the hydroxyl group of glutamine and the hydroxyl group of NADP+, and the other bond between the glutamine and the hydroxyl group of NADP+.
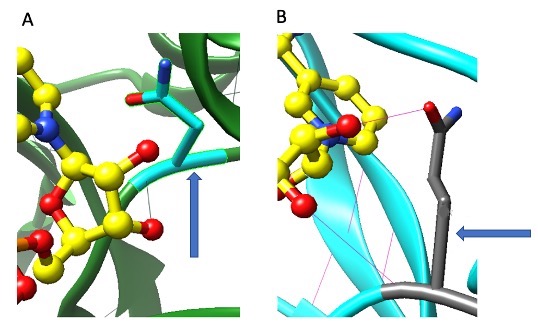
Figure 1. Structural Analysis of desired N97Q KPR mutant. The 97th amino acid of S. aureus KPR protein before mutation (A) and after mutation (B). Yellow objects are carbon molecules, red objects are oxygen molecules, and dark blue objects are nitrogen molecules. Arrow in panel A is pointing to N97 and the arrow in panel B is pointing to Q97 molecule. Faint green line in A between red dot and N97 represents hydrogen bond. Faint pink lines between red dots and Q97 molecule represent hydrogen bonds.
Designing mutagenic primers. After establishing the mutation to be created, N97Q, the next step is to design the primers that could be used for mutagenic primer-based PCR. Primers were designed using the SDM-assist [10] software program to have two mismatches. One mismatch creates a missense mutation resulting in the mutation of N97Q. The second mismatch creates a silent mutation that results in a new restriction enzyme site for the restriction endonuclease, MscI. This additional restriction enzyme site is a silent mutation but allows the verification of mutagenesis through restriction enzyme digestion analysis. The creation of a new restriction site generates a predictable and observable DNA banding pattern in an agarose gel, which allows verification. The SnapGene program (from GSL Biotech; available at snapgene.com) was used to predict the results of a restriction enzyme digestion reaction with the mutant plasmid and MscI enzyme.
Initial (failed trial) LB-Agar Plates of wildtype and mutated KPR plasmid from Experiment. The mutagenic primers were used in a PCR reaction using plasmid DNA molecule containing the gene sequence for S. aureus KPR (NCBI accession ID: NC_002758). The PCR reaction product was performed and transformed into E. coli and plated onto LB-agar plates according to the manufacturer’s recommended protocol. The results are shown in Figure 2A. We unexpectedly did not see any colonies in our experimental plate (Figure 2A, left). To understand what may have gone wrong, we looked at the results from the two controls provided with the mutagenesis kit (Figure 2A, middle and right). These controls used blue-white colony screening method to indicate successful mutagenesis and transformation. The results of these plates were identical to our experimental plate, no colonies were visible. It is hard to determine what exactly caused this type of result and there are many possible reasons involving user error such as imprecise measuring of chemical reagents or adding the wrong reagent. For example, the wrong antibiotic could have been added to each plate. This would explain why no colonies were observed even in the control reaction.
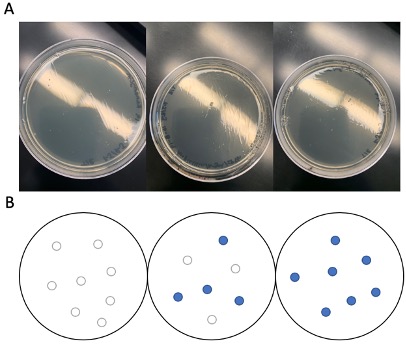
Figure 2. Transformation results of trial 1. Actual results (A) vs. theoretical/expected results (B) of experimental KPR plate (left), PCR control plate (middle), and transformation control plate (right) after the first trial of the project.
Agarose gel electrophoresis of PCR reaction (Trial 2). The initial trial (explained above) did not yield any colonies. Colony growth requires a successful PCR step and a successful transformation step. Because of the failed trial, we added an intermediate step to see if we could verify the success of the PCR reaction before moving to the transformation step. For this intermediate step, we ran an agarose gel electrophoresis assay on our PCR reaction. In theory, a successful PCR reaction would be indicated in an agarose gel with two observations- 1) a single band and 2) a single band corresponding to the expected size. With this plan in hand, a second trial was commenced with the PCR reaction as previously performed and portion of the finished reaction was assayed in an agarose gel. The results are shown in Figure 3. Looking at the gel results, a single band is visible indicating a successful PCR reaction. Although the size of this band is difficult to determine, the mere presence of this band necessarily requires a successful PCR reaction. This shows that the amplification of the DNA was a success and we can carry out the downstream steps with confidence knowing that the PCR reaction was successful.

Figure 3. Gel results of the mutagenic PCR products. Contents of each lane are as follows: Lane M- 1 kb DNA ladder, 1) KPR plasmid. The asterisk is denoting a faint band in lane 1. The size is difficult to estimate due to the streaking but was assumed to be ~6kb, which is the size of our plasmid.
Transformation of E. coli with PCR product (Trial 2). Once the success of the mutagenic PCR was confirmed, we moved forward with bacterial transformation as trial 1 but with extra attention paid to technical procedures such as reagent preparation and set-up. The results of the second trial are shown in Figure 4. Observing the results, four white colonies can be seen on the LB-agar plate. This shows a successful transformation of E. coli. Due to limited reagent availability, the control reactions were not performed, but we moved forward in the experiment nevertheless.
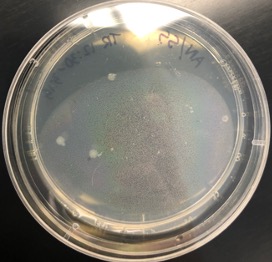
Figure 4. Transformation of E. coli with PCR product (Trial 2). Colonies are observable in the agar plate.
Verifying mutagenesis with the MscI restriction endonuclease. Now that we obtained colonies in the LB-agar plate, the next step was to determine which colony contain our mutated plasmid. A mutated plasmid could be identified after plasmid purification and restriction enzyme digestion because the primer pair used in the PCR reaction added silent mutation to create a restriction enzyme site for the restriction enzyme, MscI. The nonmutated plasmid (template DNA in PCR reaction) does not have this restriction enzyme stie. Therefore, digesting both of these samples with the MscI restriction would yield different results because the mutated plasmid could be digested but the nonmutated plasmid could not. The mutated KPR plasmid upon digestion will result in a single band in an agarose gel representing linear DNA molecule 6182bp in size. If it could not be digested (i.e. no mutation made) two bands would be visible for nonlinear conformation of plasmid (supercoiled and nicked circle conformation). The results of the digestion reaction are shown in Figure 5. A single band is visible although it is difficult to determine the exact size of this band but since only one band was detected, we interpreted this plasmid sample as being mutated.
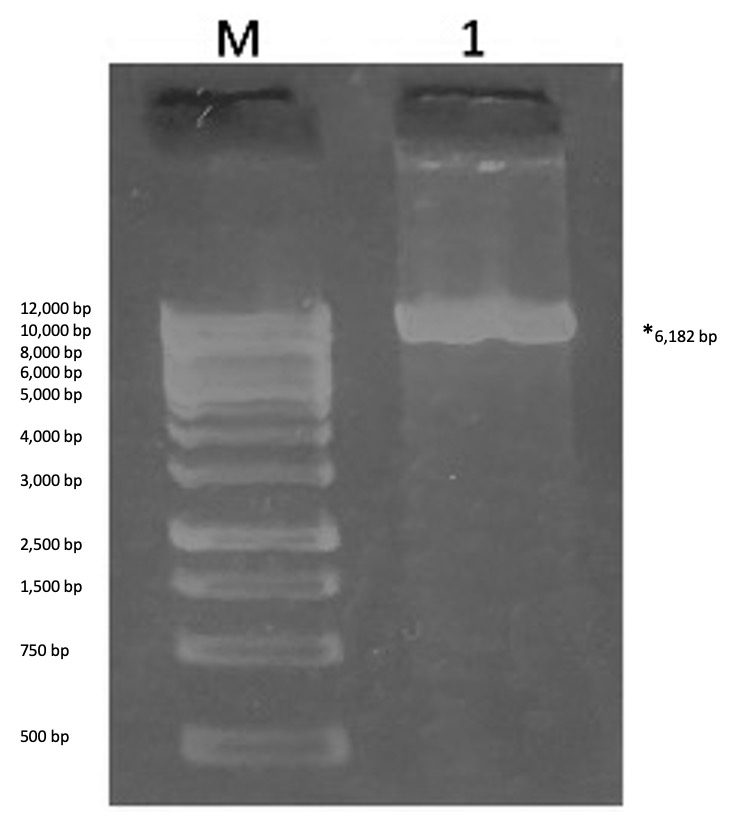
Figure 5. Agarose gel electrophoresis results for the restriction enzyme digestion assay between colony-purified KPR plasmid and Msc1. Contents of each lane are as follows: Lane M- 1 kb DNA ladder, 1) MscI-digested plasmid sample. In lane 1, a single band is visible indicating the plasmid sample was digested, resulting in a change in conformation of plasmid DNA from circular to linear.
Mutation confirmation. Restriction enzyme digestion of the purified plasmid sample served as an initial verification procedure. The sample used in lane 1, Figure 5, which showed positive indication for mutation, was sent for DNA sequencing for confirmation of mutagenesis. The sequenced DNA was analyzed with the SnapGene software program. We compared the retrieved DNA sequence and the nonmutated sequence side-by-side and the results are shown in Figure 6. The wildtype DNA codon sequence CAG (Figure 6B) was mutated into the DNA codon sequence AAT (Figure 6A) indicating we were successful in performing the missense mutation (N97Q) and the silent mutation (creation of a restriction enzyme site). It is worth mentioning that this analysis also revealed the presence of the MscI restriction enzyme site in the mutated sample as expected.
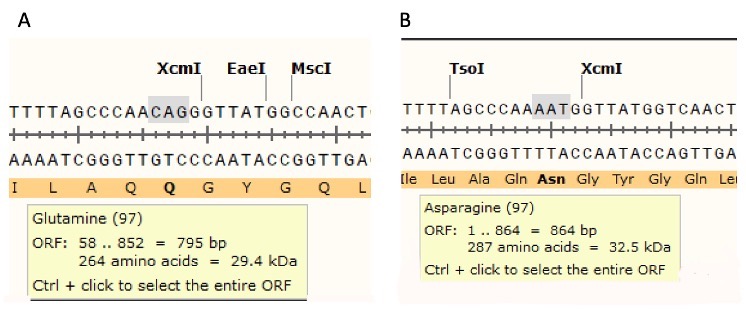
Figure 6. Experimental KPR DNA sequence with mutated Q97 and MscI restriction enzyme site (A) and wtKPR DNA sequence (B). The software program, SnapGene®, was used to analyze the sequenced data and look for presence/absence of a the MscI restriction enzyme site. The software program was capable of detecting the MscI restriction site in the DNA sample submitted or sequencing (A) but could not identify this site in the wtKPR DNA sequence. The missense mutation, N97Q is also detected.
Conclusions/Future Directions
In this article we presented the results of the initial steps of our research project with the long-term objective of structural and functional analysis of KPR Mutant. The work in this article was conducted as a classroom research project in the course BIOL3110: Basic Skills in the Laboratory. This project involves the enzyme KPR, which is essential bacterial metabolism. KPR catalyzes the reduction of Ketopantoate to Pantoate which is a precursor molecule for CoA. This coenzyme is a part of cellular respiration, so its presence is crucial for metabolism. Unlike bacteria, humans do not rely upon KPR for CoA synthesis. Because of this disparity, KPR has been identified as an anti-bacterial drug target. This means that understanding it better may give insight into designing/identifying drugs that could specifically inhibit it. Anti-bacterial agents have been found to have strong implications on disease defense in the past. One study examined the efficiency of penicillin on bacterial infections. Because of penicillin-binding proteins, an aspect of penicillin, it is efficient at inhibiting bacterial enzymes that are known to spread and cause disease. Enough insight into KPR may aid the creation of new therapeutics that may have similar antibacterial effect as penicillin.
Previous studies showed that N98 of E. coli is important for its KPR function, however, it remains to be seen whether this amino acid is important for S. aureus. In an effort to better understand this residue in S. aureus KPR, we set out on a site-directed mutagenesis experimental project to substitute this amino acid with glutamine. The project requires many steps, and we were able to successfully complete this on the DNA level by using mutagenic primers in a PCR reaction. This mutation was verified by restriction enzyme digestion and DNA sequencing. Due to the limited time available, we were not able to progress further and analyze the DNA mutation at the protein level. However, another undergraduate student could continue this work and perform mutagenesis on the protein level.
References
1. Matak-Vinkovic, D., et al., Crystal structure of Escherichia coli ketopantoate reductase at 1.7 A resolution and insight into the enzyme mechanism. Biochemistry, 2001. 40(48): p. 14493-500.
2. Begley, T.P., C. Kinsland, and E. Strauss, The biosynthesis of coenzyme A in bacteria. Vitam Horm, 2001. 61: p. 157-71.
3. Devi, P.B., et al., Structure-guided design of thiazolidine derivatives as Mycobacterium tuberculosis pantothenate synthetase inhibitors. ChemMedChem, 2014. 9(11): p. 2538-47.
4. van der Westhuyzen, R., et al., The antibiotic CJ-15,801 is an antimetabolite that hijacks and then inhibits CoA biosynthesis. Chem Biol, 2012. 19(5): p. 559-71.
5. Carter, P., Site-directed mutagenesis. Biochem J, 1986. 237(1): p. 1-7.
6. Ciulli, A., et al., Crystal structure of Escherichia coli ketopantoate reductase in a ternary complex with NADP+ and pantoate bound: substrate recognition, conformational change, and cooperativity. J Biol Chem, 2007. 282(11): p. 8487-97.
7. Center for Disease Control and Prevention. Staph infections can kill. [cited 2020 June 1 2020]; Staph infections can kill]. Available from: https://www.cdc.gov/vitalsigns/staph/index.html.
8. Sanchez, J.E., et al., Evidence of Kinetic Cooperativity in Dimeric Ketopantoate Reductase from Staphylococcus aureus. Biochemistry, 2015. 54(21): p. 3360-3369.
9. Pettersen, E.F., et al., UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem, 2004. 25(13): p. 1605-12.
10. Karnik, A., R. Karnik, and C. Grefen, SDM-Assist software to design site-directed mutagenesis primers introducing “silent” restriction sites. BMC Bioinformatics, 2013. 14: p. 105.
Acknowledgements: The data and results collected from this study and the experimentation itself was possible because of the assistance from several individuals in my course, BIOL 3110L. The experimentation was carried out in a classroom laboratory. I would first like to thank my project group members who each contributed to designing and carrying out the experiment: Kendall Jolly, Andy Nguyen, and Sherine Starr. I would also like to sincerely thank our course instructor, Dr. Jason O’Donnell, Ph.D. His constant encouragement and passion for science enabled was reassuring during times of stress and setbacks in our project. Our Graduate Learning Assistant, Luoman Chen, was also a great help during this project as she would always answer questions and give us pointers during times of confusion.