Asparagine-97 Effect on Staphylococcus aureus Ketopantoate Reductase Function in N97I Mutant Escherichia coli
by Lauren Trainer, Biology
Abstract: Staphylococcus aureus is a pathogenic bacterium that can use aerobic or anaerobic respiration for the energy synthesis required for survival and propagation via binary fission.1 Canonical bacteria exhibit a pronounced reliance on ketopantoate reductase (KPR) in the biosynthesis of energy metabolites.2 Active site modification of the KPR protein in S. aureus bacteria holds medical significance as a potential therapeutic target. Our study aimed to inhibit the production of coenzyme A via the NADPH-dependent reaction catalyzed by KPR to inhibit bacterial ATP production and induce necrotic cell death.3-5 Limited research has been conducted to study the active site of S. aureus KPR. Former research has identified the importance of Asn98 in the substrate binding capabilities on the active site of Escherichia coli KPR.6,7 Previous studies have revealed similar amino acid sequencing in S. aureus KPR, with an asparagine residue located in a near-identical position to the E. coli KPR. The structural similarities between E. coli KPR and S. aureus KPR suggest the significance of Asn97 in the active site of S. aureus KPR. To test the importance of Asn97 in S. aureus KPR, we performed site-directed mutagenesis via polymerase chain reaction (PCR) to achieve the N97I substitution. A plasmid containing the mutated S. aureus KPR gene was transferred to E. coli cells to produce the N97I mutant for analysis. Preliminary enzyme kinetics of wild-type S. aureus KPR demonstrate the dependence of S. aureus KPR on NADPH.8 Further research is required for the enzymatic analysis of mutant S. aureus KPR to identify specific therapeutic targets.
Ketopantoate reductase (KPR), Staphylococcus aureus (S. aureus), site-directed mutagenesis, enzyme kinetics
Introduction
Staphylococcus aureus is a gram-positive, facultative anaerobe presenting as individual cocci that arrange in clusters.9 S. aureus is clinically relevant due to its known cause of mammalian infection, ranging from acute dermatologic infections, such as methicillin-resistant Staphylococcus aureus (MRSA), to severe bloodstream infections, like bacteremia resulting in sepsis.9-11, 36 Treatment of S. aureus infection is complicated by increasing instances of antibiotic resistance, including resistance to methicillin, penicillin derivatives, and vancomycin.9 Antibiotic resistance to current therapies increases the demand for alternative therapeutic targets.
Ketopantoate reductase (KPR) is an essential enzyme that catalyzes the production of pantoate via the utilization of NADPH. The conversion of ketopantoate (KP) to pantoate is a crucial step in the de novo synthesis of pantothenic acid (pantothenate) (Figure 1). Pantothenic acid produced by the NADPH-dependent catalysis of KPR forms the core of coenzyme A (CoA).2, 12, 35 CoA is essential for aerobic respiration in bacterial species, functioning as a carrier of acyl groups in the formation of acetyl-CoA, a high-energy molecule that serves as a substrate for several metabolic processes.13, 14 The synthesis of pantothenic acid is essential for the growth and proliferation of pathogenic bacterial species, presenting as a possible antibiotic target.15
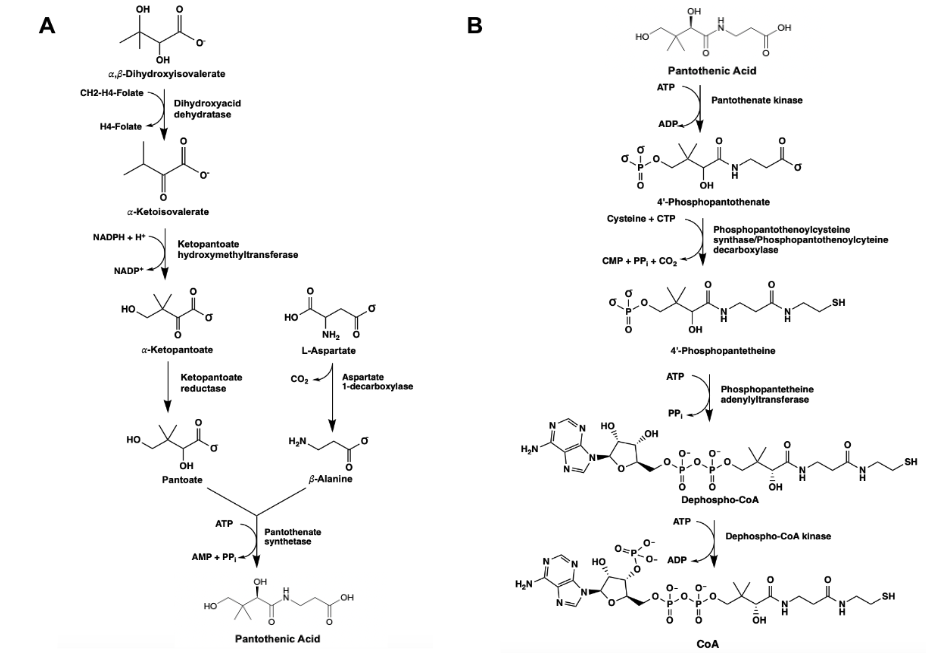
Figure 1. The de novo synthesis of pantothenic acid (pantoate) via the utilization of KPR in canonical prokaryotes (A). The biosynthetic pathway yielding CoA, succeeding the synthesis of pantoate in bacterial species (B).
Limited research has been conducted to study the catalytic mechanism of S. aureus KPR. Previous structural studies have identified E. coli KPR as a monomeric enzyme (Figure 2A) with an aperture formed by an alpha-beta fold on the N-terminal domain and eight alpha-helices on the C-terminal domain. The cofactor NADPH binds to the alpha-beta fold on the N-terminal domain. KP binds to the active site (Figure 2B) in the cleft formed at the junction between the N-terminal domain and the C-terminal domain of E. coli KPR.8, 16 Previous structural studies have identified S. aureus KPR as a dimeric enzyme bridged by hydrophobic residues (Figure 3A).8 The unique dimeric structure distinguishes S. aureus KPR from E. coli KPR.
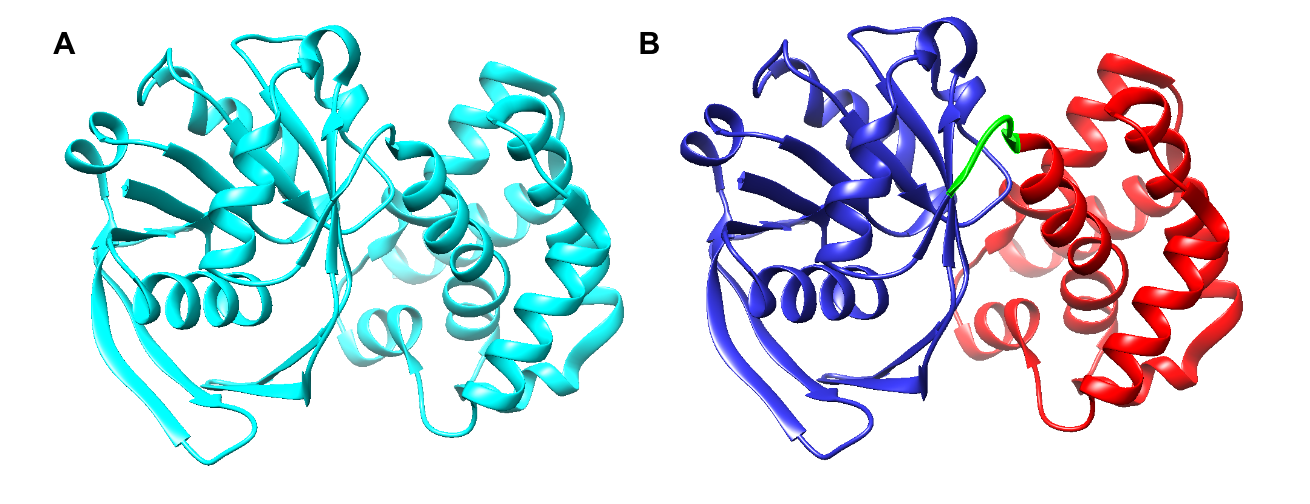
Figure 2. UCSF CHIMERA image of E. coli KPR protein structure (A) showing a monomeric arrangement of the KPR molecule (cyan) present in wild-type E. coli KPR (PDB entry 1KS9). UCSF CHIMERA image of the active site of E. coli KPR (B) showing the N-terminal (residues 1-164) (blue) and the C-terminal (residues 170-291) (red) present in wild-type E. coli KPR. KP binds to the active site in the cleft formed at the junction between the N-terminal domain and the C-terminal domain (green).
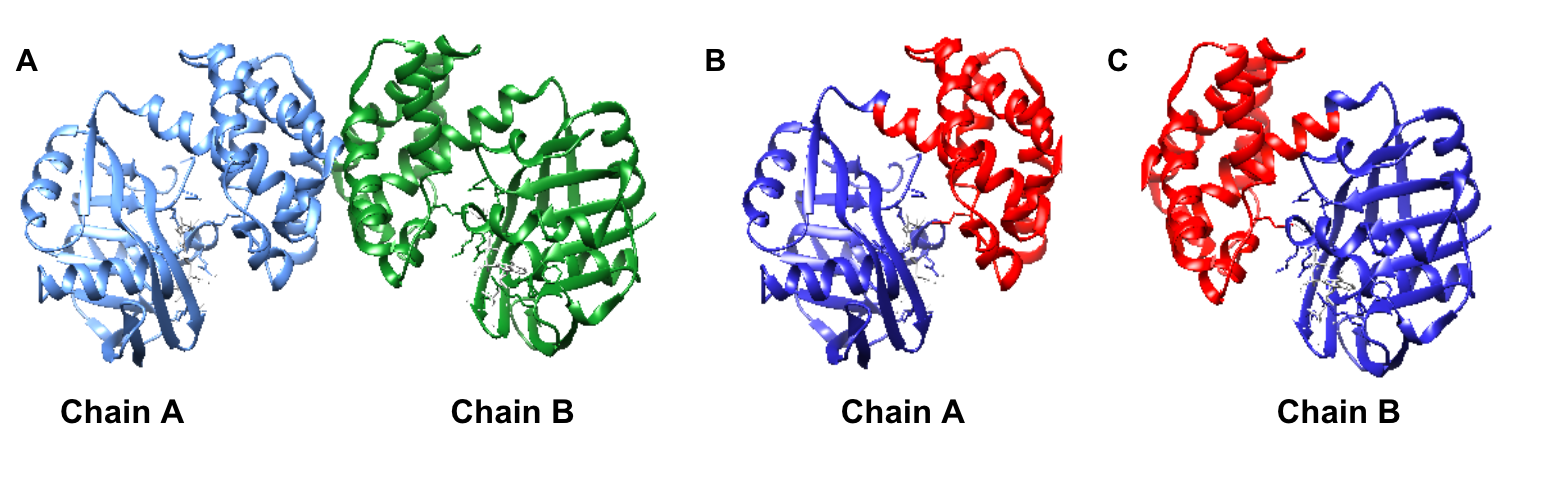
Figure 3. UCSF CHIMERA image of S. aureus KPR protein structure (A) dimeric arrangement (PDB entry 4YCA) of the two KPR molecules, chain A (blue) and chain B (green), present in wild-type S. aureus KPR. UCSF CHIMERA image of chain A of S. aureus KPR (B) showing the N-terminal (residues 1-164) (blue) and the C-terminal (residues 165-286) (red) present on chain A of wild-type S. aureus KPR. UCSF CHIMERA image of chain B of S. aureus KPR (C) showing the N-terminal (residues 1-164) (blue) and the C-terminal (residues 165-286) (red) present on chain B of wild-type S. aureus KPR. KP binds to the active site in the cleft formed at the junction between the N-terminal domain and the C-terminal domain (green).
Previous studies have identified amino acid residues and their proposed function on the active site of E. coli KPR. Arg31, Lys72, Asn98, Lys176, Asn180, Ser244, and Glu256 residues (Figure 4A) have proposed significance including pantoate interactions, substrate binding, cofactor binding, and catalytic activity on the active site of E. coli KPR.7, 17 Our study focused on the importance of Asn97 in the S. aureus KPR gene. The Asn97 residue was selected due to its structural similarity to Asn98 in E. coli KPR. Asn98 is a residue present on the active site that binds KP in E coli KPR. The near-identical position and sequencing of the Asn98 and Asn97 residues present on E. coli KPR and S. aureus KPR suggest the importance of Asn97 in S. aureus KPR function (Figure 4). Asparagine is a hydrophilic amino acid located on the outer domain of globular proteins (Figure 4B). Asn97 was mutated to isoleucine, a hydrophobic amino acid located in the core of globular proteins (Figure 4C). This critical change in the hydrophobicity of the 97th amino acid aimed to alter the conformation of the active site of the S. aureus KPR protein, subsequently affecting the binding capabilities of NADPH.
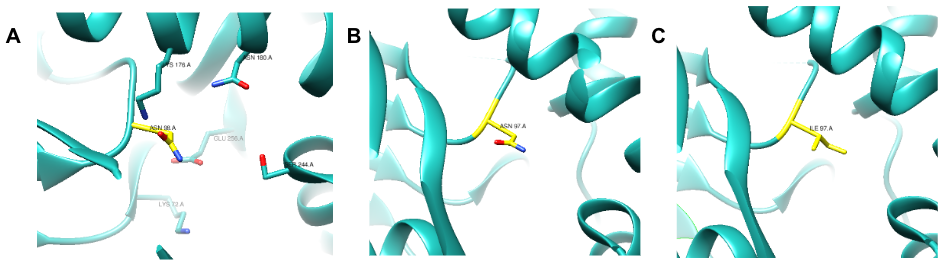
Figure 4. UCSF CHIMERA image of E. coli KPR (cyan) (PDB entry 1KS9) protein structure (A) showing the stick structure of amino acid residues present on the active site of wild-type E. coli KPR (A) including Asn98 (yellow). UCSF CHIMERA image of S. aureus KPR (cyan) (PDB entry 4YCA) protein structure (B) showing the stick structure of Asn97 present on chain A ofwild-type S. aureus KPR (yellow). UCSF CHIMERA image of mutant S. aureus KPR (cyan) protein structure (C) showing the stick structure of Ile97 (yellow) present on chain A of the N97I mutant S. aureus KPR. The visible amino acid residues are colored by heteroatom.
Methods
To make the protein of interest, we used a plasmid containing the S. aureus KPR gene that was gifted to us by Dr. Zachary Wood, Professor in the Department of Biochemistry and Molecular Biology at the University of Georgia.
Mutagenic Polymerase Chain Reaction (PCR)
Polymerase chain reaction (PCR) was selected to perform site-directed mutagenesis due to its high sensitivity and reproducibility.18 PCR allows for the targeted introduction of specific mutations at precise locations within a gene via oligonucleotide primers. The selective mutations performed by PCR can alter protein-protein interactions, posttranslational modifications, or enzymatic activity.19 Our study mutated the 97th amino acid of the S. aureus KPR gene from asparagine to isoleucine.
Transformation
To determine if Asn97 is important for the function of S. aureus KPR we first needed to express N97I mutant E. coli cells containing the mutated S. aureus KPR gene of interest. The mutated S. aureus KPR gene was transformed into a non-pathogenic strain of E. coli, BL21(DE3) to produce the N97I mutant. The plasmid transferred into ultracompetent E. coli cells encoded genes for the mutant S. aureus KPR protein, kanamycin resistance (KanR), and the lac operon promoter (lacI).
Protein Expression
Kanamycin was added to the agar plates at a final concentration of 50 µg/ml to select for bacteria that contained the plasmid of interest, the N97I mutant, and to select against bacteria that did not contain the plasmid of interest, E. coli cells that were unsuccessful in the transformation of the plasmid or alternative bacterial contaminants. The N97I mutant was streaked onto 50 µg/ml kanamycin-containing 1.0% agar and incubated for 1-2 days at 37 °C to achieve individual colonization. An isolated bacterial colony of the N97I mutant strain was inoculated into 5 ml of 50 µg/ml kanamycin-containing lysogeny broth (LB) and incubated for 1-2 days at 37ºC. The inoculation culture was scaled up via a 1:20 dilution with 50 ml of LB media and was shaken at 200 rpm for 3 hours at 37ºC.
To determine if Asn-97 is important for S. aureus KPR function it was essential to express the mutant protein and purify it. The mutant S. aureus KPR gene operates under the Lac Operon expression vector system inducible by Isopropyl ß-D-1-thiogalactopyranoside (IPTG).20, 21 Because of its regulation of lac promoter transcription via the mimicry of allolactose, IPTG was selected to induce the expression of the mutated S. aureus KPR gene at a final concentration of 0.42 mM.22 IPTG induced protein expression following the inoculation of the N97I mutant into growth media at 20ºC shaken at 80 rpm for 18 hours.23
Protein Purification
To obtain N97I mutant protein samples for analysis, the harvested cells were lysed and purified. Cells were first harvested by centrifugation at 6,000 rpm for 2 minutes. The pellets containing the harvested cells of interest were combined with a lysis buffer containing 50 mM sodium phosphate and 300 mM sodium chloride at a neutral pH of 7.8. Ultrasonication via a sonicator probe was preferred to disrupt the membrane of harvested N97I mutant cells via the alteration of tertiary and secondary protein structure, resulting in cell lysis.24 Sonication was performed in 1-second intervals over ice for a total duration of 5 minutes to sonicate 10 ml of solution. To isolate the protein of interest, the sonicated cells were centrifuged at 14,800 rpm for 20 minutes. The supernatant of the centrifuged sample was purified to collect the isolated protein.
Affinity chromatography was preferred for purifying the harvested protein samples due to its exceptional recovery yield and purity levels for isolating recombinant proteins containing His-tags.2, 25 Protein purification by immobilized metal affinity chromatography (IMAC) with TALON resin was performed to isolate and purify the mutant S. aureus KPR protein. TALON resin is a cobalt-containing chelating agent that binds specifically to His-tagged proteins, allowing for their selective purification.26 The S. aureus KPR protein of interest contains a His-tag at the C-terminus.8, 16 The IMAC apparatus was prepared with 0.5 ml of TALON resin 5 ml of lysis buffer containing 50mM sodium phosphate, and 300mM of NaCl prior to protein purification. The affinity column was washed with 7 ml of a 7.8 pH wash buffer containing 50mM sodium phosphate, 300 mM NaCl, and 25 mM imidazole followed by the addition of 4ml of 7.8 pH elution buffer containing 50 mM sodium phosphate, 300mM NaCl, and 150 mM imidazole to purify and collect the protein of interest.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
To proceed with enzymatic analysis it was essential to verify the presence of the purified protein of interest. The relative presence of KPR recombinant protein expression was detected using analytical sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie Blue staining (Figure 8).27 SDS PAGE gel electrophoresis was selected to resolve the fractioned protein samples for its timely efficient qualitative and semiquantitative estimation of molecular weight.28 Each fraction of the purified protein sample was combined with an equal volume of 2X loading buffer; 15 µl of each fraction sample and 15 µl of loading buffer were incubated at 95ºC for 10 minutes. The incubated samples were loaded into the gel and SDS PAGE gel electrophoresis was performed at 200V for 30min with a 1X TGS (Tris-Glycine-SDS) buffer.
G-250 Dye-binding (Bradford) Protein Assay
The concentration of protein present in dialyzed samples of the purified protein eluted from affinity chromatography was quantified by a Coomassie blue G-250 dye-binding (Bradford) protein assay.29 The purified protein was dialyzed and further purified via a Slide-a-lyzer mini dialysis device by ThermoScientific™ and a 7.5 pH dialysis buffer containing 25 mM tris and 50 mM NaCl prior to quantification. A polypropylene membrane was selected for protein dialysis due to its high recovery of large proteins (greater than 10 kDa).30 The S. aureus KPR protein of interest is 34kDa in its monomeric denatured state yielded by sonication. Serial dilutions were performed on the dialyzed protein samples and Bovine Serum Albumin (BSA) samples for the generation of a standard curve triplicate and dialyzed protein duplicate via a Bradford protein assay. A Bradford protein assay was preferred for the quantification of protein concentration due to reproducibility and rapid colorization.31 Absorbance values of the standard curve and dialyzed protein samples were measured at 595 nm and 450 nm and a linearization of the Bradford protein assay was performed.32
Enzyme Kinetics
Functional analysis of the S. aureus KPR protein was performed through enzyme kinetics via ultraviolet-to-visible (UV-Vis) spectroscopy. UV-Vis spectroscopy was selected for the analysis of the S. aureus KPR protein due to its quantitative estimation of absorbance and qualitative estimation of the progression of enzymatic reactions.33 Equal volumes of each reagent including the protein sample, 150mM NADPH, and 300mM KP were mixed in a 100 µl reaction vessel for 100 seconds. During the reaction progression 100 scans were collected; the absorbance values at 340 nm, corresponding to the wavelength of NADPH, were used to generate a progress curve.
UCSF CHIMERA
UCSF CHIMERA was used to generate images of the E. coli and S. aureus KPR proteins of interest using experimentally determined biomolecular structures from the Worldwide Protein Data Bank (wwPDB).37, 38 The monomeric conformation of E. coli KPR (PDB entry 1KS9), the dimeric conformation of E. coli (PDB entry 2OFP), and the dimeric conformation of S. aureus KPR (PDB entry 4YCA) were visualized using UCSF CHIMERA.34
Results
Protein Expression
The growth of the N97I mutant on kanamycin-containing agar plates indicates the successful transfer of the mutated S. aureus plasmid into E. coli cells. The E. coli cells that successfully transformed the S. aureus gene of interest transformed the gene for kanamycin resistance in addition to the mutated KPR gene. A colony of genetically identical bacteria cells that successfully transformed the mutant plasmid of interest was selected for further inoculation and protein expression.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS PAGE)
The fractions collected throughout protein purification were used to determine the relative presence of KPR recombinant protein expression using analytical SDS PAGE and Coomassie Blue staining (Figure 5). Lane 1 indicates a sample of clarified lysate collected before protein purification. Lanes 2-3 indicate fractions of the flow through collected after the addition of a wash buffer. Lanes 2-3 contained impurities, components that did not bind to the TALON resin, and the components of the wash buffer (sodium phosphate, sodium chlorate, and imidazole). Decreasing concentrations of impurities were present in each sample collected as the buffer progressed through the column. Lanes 4-6 indicate fractions of the flow through collected after the addition of an elution buffer. Lanes 4-6 contained the target protein, minimal impurities, wash buffer components, and elution buffer components (sodium phosphate, sodium chlorate, and imidazole). We found that lane 4 contained significant quantities of the target protein, outlined by the visible bands present in lane 4.
Decreasing concentrations of impurities were present in each sample collected as the buffer progressed through the column. The greatest concentration of the target protein was observed in lane 4, containing the first elution fractions. The molecular weights of the fractioned protein samples were analyzed via comparison to the markers generated by the 26619 Thermo Scientific™ PageRuler™ Plus Prestained Protein Ladder (10 to 250 kDa).3 The SDS PAGE indicated the successful purification of the S. aureus KPR protein of interest. The elution fractions corresponding to the highest concentration of target protein estimated the molecular weight of 34kDa as the molecular weight of the purified protein, corresponding to the molecular weight of monomeric S. aureus KPR.4
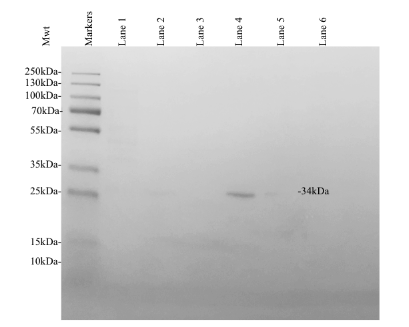
Figure 5. Coomassie Blue stained analytical SDS PAGE showing the expression of recombinant S. aureus KPR. Lane 1 was loaded with a sample of clarified lysate collected prior to protein purification. Lanes 2-3 were loaded with a sample of flow through both immediately following and after the addition of a wash buffer. Lanes 4-6 were loaded with a sample of flow through both immediately following and after the addition of an elution buffer. Thermo Scientific™ PageRuler™ Plus Prestained Protein Ladder, 10 to 250 kDa was loaded as markers. The expression of a 34 kDa protein was observed, corresponding to the expected molecular weight of His-tagged recombinant monomeric KPR.
G-250 Dye-binding (Bradford) Protein Assay
The relative concentration of the mutant protein of interest was quantified via a Bradford protein assay. The absorbance values collected at 595 nm and 450 nm were used to generate a standard curve (Figure 6). The standard curve included known concentrations of BSA (0 µg/ml, 15 µg/ml, 30 µg/ml, 45 µg/ml, and 60 µg/ml) indicated by the dashed line (Figure 6). Diluted samples of the N97I mutant KPR protein were compared to known BSA concentrations to determine the concentration of N97I mutant KPR protein present in the sample collected. The Bradford protein assay estimated the concentration of N97I mutant KPR protein collected to be 9.196 µg/ml.
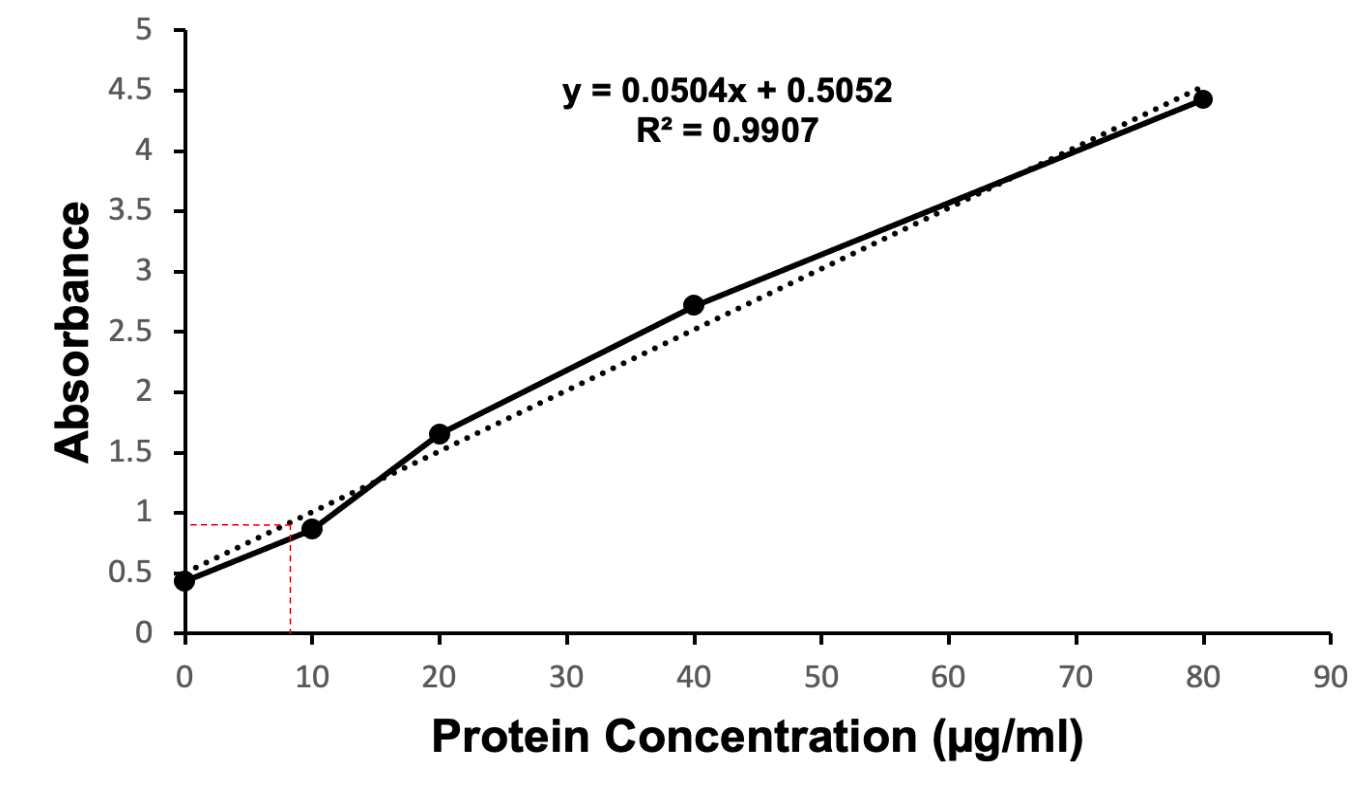
Figure 6. Coomassie Brilliant Blue stained quantitative Bradford protein assay showing the estimated coordinates identifying the concentration of recombinant S. aureus KPR protein (dashed line in red). Concentrations of BSA (0 µg/ml, 15 µg/ml, 30 µg/ml, 45 µg/ml, 60 µg/ml) were used to generate a standard curve (dashed line in black). Diluted samples of the N97I mutant KPR protein were compared to the standard curve to determine the concentration of N97I mutant KPR protein present in the sample collected. The Bradford protein assay estimated the concentration of N97I mutant KPR protein harvested to be 9.196 µg/ml.
Enzyme Kinetics
Wild-type S. aureus KPR was used to study the relationship between S. aureus KPR and NADPH. Enzymatic analysis of wild-type S. aureus KPR was performed via (UV-Vis) spectroscopy. UV-Vis spectroscopy was selected for the analysis of the S. aureus KPR protein due to its quantitative estimation of absorbance and qualitative estimation of the progression of enzymatic reactions.33 Preliminary enzyme kinetics of wild-type S. aureus KPR demonstrate the dependence of S. aureus KPR on NADPH (Figure 7).
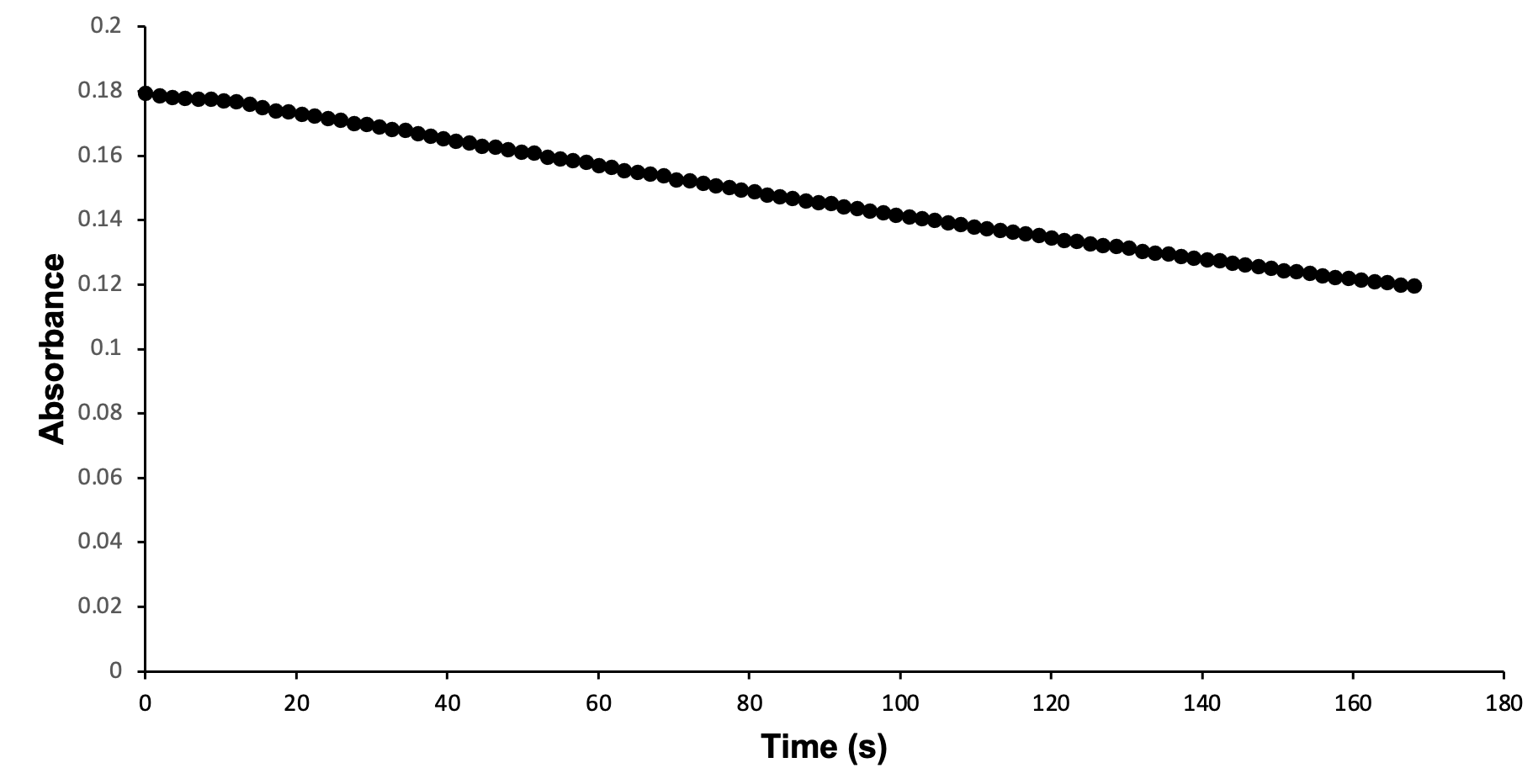
Figure 7. Enzymatic activity of wild-type S. aureus KPR produced via UV-Vis spectroscopy. The absorbance of NADPH was measured at 340 nm, showing the progression of a reaction containing 33 µl of wild-type S. aureus KPR, 150 mM NADPH, and 300 mM KP. The reaction proceeded for 100 seconds prior to scan collection. 100 scans were used to generate a progress curve.
Discussion
Preliminary analysis of enzyme kinetics confirms the dependence of wild-type S. aureus KPR on NADPH during the enzymatic reaction of purified wild-type S. aureus KPR protein, NADPH, and KP. The decreasing absorbance values observed during the reaction progression correspond to decreasing concentrations of NADPH, indicating the consumption of NADPH during the reaction of purified wild-type S. aureus KPR protein, NADPH, and KP. The reliance of S. aureus KPR on NADPH suggests active-site modification of the KPR protein as an antibiotic target. Successful alteration of the active site is expected to inhibit the binding of KP to the active site of KPR. The NADPH-dependent reduction of KP would not proceed without successful binding at the active site, preventing the consumption of NADPH in the reaction of mutant S. aureus KPR protein, NADPH, and KP. The NADPH consumed in the reaction involving mutant KPR would remain stagnant, corresponding to stationary absorbance values that remain high in the enzymatic analysis of mutant S. aureus KPR.
The study performed was an in vitro study of isolated protein harvested from lysed cells combined with stock solutions of KP and NADPH. The study was limited by the absence of live bacterial cells during the reaction progression. An in vivo study to assess the effects of mutant KPR protein in a live cellular model would progress the study to identify the effects of mutant KPR in the metabolic capabilities of live S. aureus bacteria.
Experiments targeting alternative conserved residues between bacterial species could reveal metabolic significance and identify amino acids present on the active site of S. aureus KPR, further identifying therapeutic targets for the treatment of infections complicated by S. aureus bacteria. This research presents potential therapeutic targets in the pantothenate biosynthesis pathway. Active-site modification of the essential enzymes required for the de novo synthesis of pantothenate in prokaryotes presents alternative antibiotic targets, including dihydroxy-acid dehydratase, ketopantoate hydroxymethyltransferase, and pantothenate synthetase. The identification of novel therapeutic targets is necessary to combat the increasing antibiotic resistance of infections complicated by S. aureus bacteria.
References
- Hammer, N. D. Renier, M.L. Cassat, J.E. Zhang, Y. Hirsch, A.O. Hood, M.I. Skaar, E.P. Two heme-dependent terminal oxidases power staphylococcus aureus organ-specific colonization of the vertebrate host. mBio 4, (2013).
- Miller, C. N., LoVullo, E. D., Kijek, T. M., Fuller, J. R., Brunton, J. C., Steele, S. P., Taft-Benz, S. A., Richardson, A. R., & Kawula, T. H. PanG, a new ketopantoate reductase involved in pantothenate synthesis. J. Bacteriol. 195, 965–976 (2013)
- Zamaraeva, M. V. Sabirov, R. Z., Maeno, E., Bessonova, S. V., & Okada, Y. Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ. 12, 1390–1397 (2005).
- Leist, M., Single, B., Castoldi, A. F., Kühnle, S. & Nicotera, P. Intracellular Adenosine Triphosphate (ATP) Concentration: A Switch in the decision between apoptosis and necrosis. J. Exp. Med. 185, 1481–1486 (1997).
- Eguchi, Y., Shimizu, S. & Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 57, 1835–1840 (1997).
- Lobley, C. M. C. Ciulli, A. Whitney, H.M. Williams, G. Smith, A.G. Abell, C. Blundell, T.L. The crystal structure of Escherichia coli ketopantoate reductase with NADP+ bound. Biochemistry 44, 8930–8939 (2005).
- Ciulli, A., Chirgadze, D. Y., Smith, A. G., Blundell, T. L. & Abell, C. Crystal structure of Escherichia coli ketopantoate reductase in a ternary complex with NADP+ and pantoate bound: substrate recognition, conformational change, and cooperativity. J. Biol. Chem. 282, 8487–8497 (2007).
- Sanchez, J. E., Gross, P. G., Goetze, R. W., Peeples, W. B., & Wood, Z. A. Evidence of kinetic cooperativity in dimeric ketopantoate reductase from Staphylococcus aureus. Biochemistry 54, 3360–3369 (2015).
- Taylor, T. A. & Unakal, C. G. Staphylococcus aureus infection. in StatPearls, (2023).
- Kwiecinski, J. M., & Horswill, A. R. Staphylococcus aureus bloodstream infections: Pathogenesis and regulatory mechanisms. Curr. Opin. Microbiol. 53, 51-60 (2020).
- C. Cheung, G. Y., Bae, J. S., & Otto, M. (2021). Pathogenicity and virulence of Staphylococcus aureus. Virulence 12, 547-569 (2021).
- Leonardi, R., & Jackowski, S. Biosynthesis of pantothenic acid and coenzyme A. EcoSal Plus. 2, (2007).
- Czumaj, A., Szrok-Jurga, S., Hebanowska, A., Turyn, J., Swierczynski, J., Sledzinski, T., & Stelmanska, E. The pathophysiological role of coA. Int. J. Mol. Sci. 21, (2020).
- Begley, T. P., Kinsland, C. & Strauss, E. The biosynthesis of coenzyme A in bacteria. Vitam. Horm. 61, 157–171 (2001).
- Spry, C., Kirk, K. & Saliba, K. J. Coenzyme A biosynthesis: an antimicrobial drug target. FEMS Microbiol. Rev. 32, 56–106 (2008).
- Matak-Vinković, D. Matak-Vinković, M. Sakdanha, S.A. Ashurst, J.L. von Delft, F. Inoue, T. Miguel, R.N. Smith, A.G. Blundell, T.L. Abell, C. Crystal structure of Escherichia coli ketopantoate reductase at 1.7 A resolution and insight into the enzyme mechanism. Biochemistry 40, 14493–14500 (2001).
- Zheng, R. & Blanchard, J. S. Kinetic and mechanistic analysis of the E. coli panE-encoded ketopantoate reductase. Biochemistry 39, 3708–3717 (2000).
- Mullis, K. B. & Faloona, F. A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 155, 335–350 (1987).
- Bachman, J. Site-directed mutagenesis. Methods Enzymol. 529, 241–248 (2013).
- Parker, A. C. & Smith, C. J. Development of an IPTG inducible expression vector adapted for bacteroides fragilis. Plasmid 68, 86–92 (2012).
- Simas, R. G., Pessoa Junior, A. & Long, P. F. Mechanistic aspects of IPTG (isopropylthio-β-galactoside) transport across the cytoplasmic membrane of Escherichia coli-a rate limiting step in the induction of recombinant protein expression. J. Ind. Microbiol. Biotechnol. 50, (2023).
- Wheatley, R. W., Lo, S., Jancewicz, L. J., Dugdale, M. L. & Huber, R. E. Structural explanation for allolactose (lac operon inducer) synthesis by lacZ β-Galactosidase and the evolutionary relationship between allolactose synthesis and the lac repressor. J. Biol. Chem. 288, 12993–13005 (2013).
- Briand, L., Marcion, G., Kriznik, A., Heydel, J. M., Artur, Y., Garrido, C., Seigneuric, R., & Neiers, F. A self-inducible heterologous protein expression system in Escherichia coli. Sci. Rep. 6, (2016).
- Jin, J., Okagu, O. D., Yagoub, A. E. A. & Udenigwe, C. C. Effects of sonication on the in vitro digestibility and structural properties of buckwheat protein isolates. Ultrason. Sonochem. 70, (2020).
- Pina, A. S., Lowe, C. R. & Roque, A. C. A. Challenges and opportunities in the purification of recombinant tagged proteins. Biotechnol. Adv. 32, 366–381 (2014).
- Chen, X., Nomani, A., Patel, N., & Hatefi, A. Production of low-expressing recombinant cationic biopolymers with high purity. Protein Expr. Purif. 134, 11-17 (2017).
- Gabe, C. M., Brookes, S. J. & Kirkham, J. Preparative SDS PAGE as an alternative to His-Tag purification of recombinant amelogenin. Front. Physiol. 8, (2017).
- Nowakowski AB, Wobig WJ, Petering DH. Native SDS-PAGE: high resolution electrophoretic separation of proteins with retention of native properties including bound metal ions. Metallomics 6, 1068-1078 (2014).
- Brady, P. N. & Macnaughtan, M. A. Evaluation of colorimetric assays for analyzing reductively methylated proteins: biases and mechanistic insights. Anal. Biochem. 491, 43–51 (2015).
- Tomczak, W. & Gryta, M. Comparison of polypropylene and ceramic microfiltration membranes applied for separation of 1,3-PD fermentation broths and Saccharomyces cerevisiae yeast suspensions. Membranes 11, (2021).
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248-254 (1976).
- Ernst, O. & Zor, T. Linearization of the bradford protein assay. J. Vis. Exp. 38, (2010).
- Worsfold, P.J. Spectrophotometry. Ency. Anal. Sci. 2, (2005).
- Pettersen, E.F. Goddard, T.D. Huang, C.C. Couch, G.S. Greenblatt, D.M. Meng, E.C. Ferrin, T.E. UCSF Chimera – A visualization system for exploratory research and analysis. J. Comp. Chem. 25, 1605-1612 (2004).
- López-Sámano, M., Lozano-Aguirre Beltrán, L. F., Sánchez-Thomas, R., Dávalos, A., Villaseñor, T., & García-García, J. D. A novel way to synthesize pantothenate in bacteria involves β-alanine synthase present in uracil degradation pathway. MicrobiologyOpen 9, 2020.
- Mulligan, M. E., Murray-Leisure, K. A., Ribner, B. S., Standiford, H. C., John, J. F., Korvick, J. A., Kauffman, C. A., & Yu, V. L. Methicillin-resistant Staphylococcus aureus: A consensus review of the microbiology, pathogenesis, and epidemiology with implications for prevention and management. Am. J. Med. 94, 313-328 (1993).
- Burley, S.K. Bhikadiya, C, Bi. C. Bittrich, S. Chao, H. Chen, L. Craig, P.A. Crichlow, G.V. Dalenberg, K. Duarte, J.M. Dutta, S. Fayazi, M. Feng, Z. Flatt, J.W. Ganesan, S.J. Ghosh, S. Goodsell, D.S. Green, R.K. Guranovic, V. Henry, J. Hudson, B.P. Khokhriakov, I. Lawson, C.L. Liang, Y. Lowe, R. Peisach, E. Persikova, I. Piehl, D.W. Rose, Y. Sali, A. Segura, J. Sekharan, M. Shao, C. Vallat, B. Voigt, M. Webb, B. Westbrook, J.D. Whestone, S. Young, J.Y. Zalevsky, A. Zardecki, C. RCSB Protein Data bank: Tools for visualizing and understanding biological macromolecules in 3D. Protein Sci. 31, (2022).
- Berman, H., Henrick, K. & Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 10, 980 (2003).
Acknowledgements: I would like to sincerely thank our course instructor and mentor, Dr. Jason O’Donnell. I am especially grateful for his continued guidance and encouragement throughout this project. I would also like to thank Dr. Zachary Wood for providing the topic of interest and the plasmid containing our mutated gene of interest. The work in this lab would not have been possible without the work of Drs. O’Donnell and Wood, and the previous work of biology students in addition to my lab members at the University of Georgia.
Citation Style: Nature